

2023年4月(yuè)9日,安徽醫科大(dà)學/南(nán)京醫科大(dà)學孫倍成團隊在Journal of Hepatology(If=30)在線發表了(le)題爲“A TNFα/Miz1-positive feedback loop inhibits mitophagy in hepatocytes and propagates nonalcoholic steatohepatitis”的(de)研究論文,該研究發現在人(rén)類NASH中,Miz1的(de)含量在肝細胞中減少。Miz1被證明(míng)與過氧化(huà)物(wù)還(hái)蛋白6 (PRDX6)結合,在細胞質中能阻止PRDX6在Cys431位點與線粒體Parkin相互作用(yòng),促進Parkin介導的(de)線粒體自噬。該研究結果表明(míng),在NASH肝髒中肝細胞Miz1的(de)缺失會導緻PRDX6抑制線粒體自噬,肝細胞中功能失調的(de)線粒體增加,以及肝巨噬細胞産生促炎細胞因子,包括TNFα。

一、研究背景
非酒精性脂肪性肝炎(NASH)是一種慢(màn)性炎症性疾病,可(kě)進一步發展爲肝硬化(huà)和(hé)肝細胞癌。然而,這(zhè)一過程背後的(de)關鍵分(fēn)子機制尚不清楚。安徽醫科大(dà)學第一附屬醫院孫倍成團隊通(tōng)過RNA測序和(hé)液相色譜-質譜分(fēn)析了(le)人(rén)類NASH和(hé)正常肝組織樣本,确定肝細胞胞漿蛋白Miz1是NASH進展的(de)潛在靶點。作者用(yòng)肝細胞特異性Miz1基因敲除和(hé)過表達小鼠建立了(le)NASH模型。通(tōng)過人(rén)NASH肝髒類器官證實其機制,免疫沉澱和(hé)質譜法用(yòng)來(lái)檢測可(kě)能與Miz1相互作用(yòng)的(de)蛋白。
二、主要結果
1. 脂肪肝中Miz1蛋白降解與NAFLD的(de)嚴重程度相關
爲了(le)說明(míng)Miz1在人(rén)類NASH中的(de)臨床意義,作者用(yòng)質譜法檢測了(le)Miz1在人(rén)類NASH (n = 5)和(hé)正常組織(n = 5)中的(de)表達,結果顯示Miz1在NASH中顯著降低。單純脂肪變性患者肝髒中的(de)Miz1蛋白水(shuǐ)平低于非脂肪變性對(duì)照(zhào)組,而在NASH組中更低(圖1A-C)。此外,肝髒中Miz1蛋白水(shuǐ)平與NASH特征呈負相關,活動評分(fēn)、體重指數和(hé)生化(huà)标準都證明(míng)了(le)這(zhè)一點(圖1D)。此外,在WDF(添加15%果糖的(de)西式飲食)和(hé)MCDD(缺乏蛋氨酸和(hé)膽堿飲食)誘導的(de)NASH小鼠模型中也(yě)證實了(le)人(rén)類Miz1數據的(de)直接相關性(圖1E)。
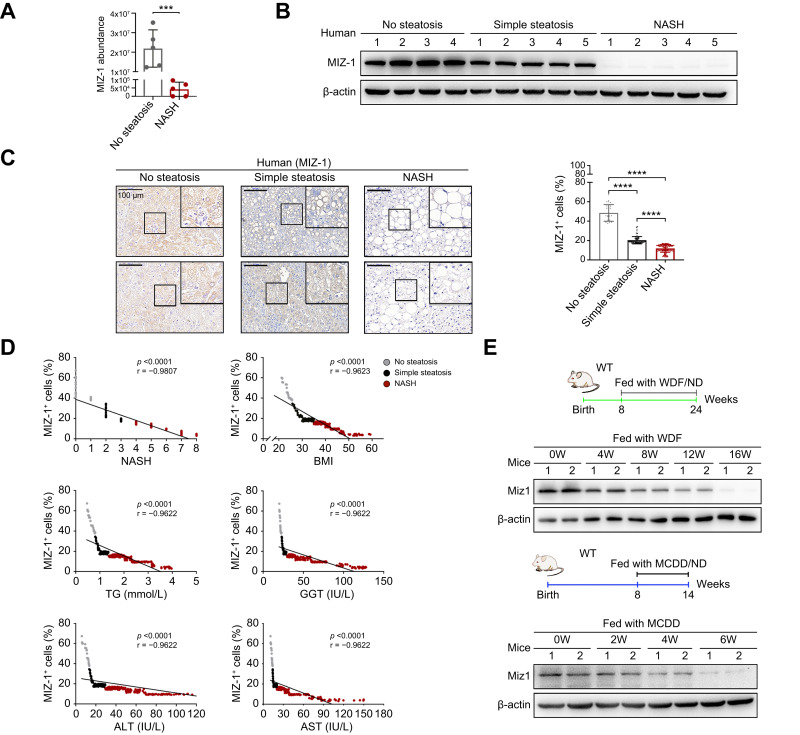
2.肝細胞特異性Miz1缺乏可(kě)促進NASH的(de)發展
爲了(le)研究Miz1在NASH中的(de)作用(yòng),作者構建了(le)肝細胞特異性Miz1全長(cháng)缺失和(hé)Miz1(POZ)敲除小鼠(以下(xià)簡稱ΔHep或POZΔHep)。五組小鼠(野生型[WT]、POZF/F、POZΔHep、F/F和(hé)ΔHep)分(fēn)别飼喂WDF或MCDD誘導NASH(圖2A)。所有小鼠NASH模型均顯示肝細胞空泡化(huà)、脂肪變性和(hé)纖維化(huà),其他(tā)四組之間無顯著差異。血清生化(huà)水(shuǐ)平、循環丙氨酸轉氨酶/天冬氨酸轉氨酶和(hé)NASH标志物(wù)也(yě)與這(zhè)些發現一緻(圖2B, D)。Miz1缺失引起的(de)變化(huà)在肝細胞特異性Miz1(POZ)敲除小鼠中不存在,表明(míng)它們不是由于Miz1的(de)轉錄調節。采用(yòng)注射AAV8-Miz1或Miz1(ΔPOZ)的(de)WT小鼠建立NASH模型。結果表明(míng),肝細胞中Miz1的(de)過表達可(kě)以獨立于POZ結構域減少NASH的(de)發展(圖2E-H)。總的(de)來(lái)說,研究結果證明(míng)在人(rén)類NASH肝髒中看到的(de)較低的(de)Miz1不是偶然的(de)相關性,Miz1的(de)缺失是NASH發展的(de)原因,這(zhè)與它的(de)轉錄活性無關。

3. Miz1缺乏導緻線粒體自噬的(de)抑制和(hé)受損線粒體的(de)積累
通(tōng)過透射電鏡(TEM)檢查了(le)小鼠肝組織中的(de)線粒體。Miz1缺失導緻線粒體腫脹積聚,嵴結構異常(圖3A)。從F/F和(hé)ΔHep小鼠中提取原代肝細胞(以下(xià)簡稱F/F和(hé)ΔHep原代肝細胞),并用(yòng)遊離脂肪酸(FFAs)刺激以模拟體内代謝應激的(de)影(yǐng)響。免疫熒光(guāng)顯示Miz1可(kě)以影(yǐng)響線粒體積累,而耗氧比和(hé)細胞外酸化(huà)率分(fēn)析顯示Miz1不參與調節線粒體功能。這(zhè)些結果表明(míng),Miz1可(kě)能與線粒體清除有關,而不是與線粒體代謝的(de)變化(huà)有關。
作者通(tōng)過對(duì)線粒體蛋白進行免疫印迹分(fēn)析,結果表明(míng)Miz1缺失導緻線粒體蛋白積累,包括泛素結合支架蛋白p62。與其他(tā)三組相比,ΔHep組自噬标志物(wù)LC3-II/I比值降低(圖3B)。然後分(fēn)離F/F和(hé)ΔHep原代肝細胞,用(yòng)原載體羰基氰化(huà)間氯苯基肼(CCCP)處理(lǐ),誘導線粒體自噬。這(zhè)些體外實驗證實了(le)體内研究結果(圖3C)。此外,Miz1缺乏會産生過多(duō)的(de)線粒體ROS,表明(míng)受損線粒體的(de)積累(圖3D)。此外,免疫熒光(guāng)顯示ΔHep的(de)線粒體自噬通(tōng)量明(míng)顯低于F/F原代肝細胞(圖3E)。通(tōng)過抗Tomm20免疫熒光(guāng)顯微鏡發現ΔHep原代肝細胞線粒體清除率不足(圖3F)。這(zhè)些數據表明(míng)Miz1缺乏導緻線粒體自噬的(de)抑制和(hé)受損線粒體的(de)積累。
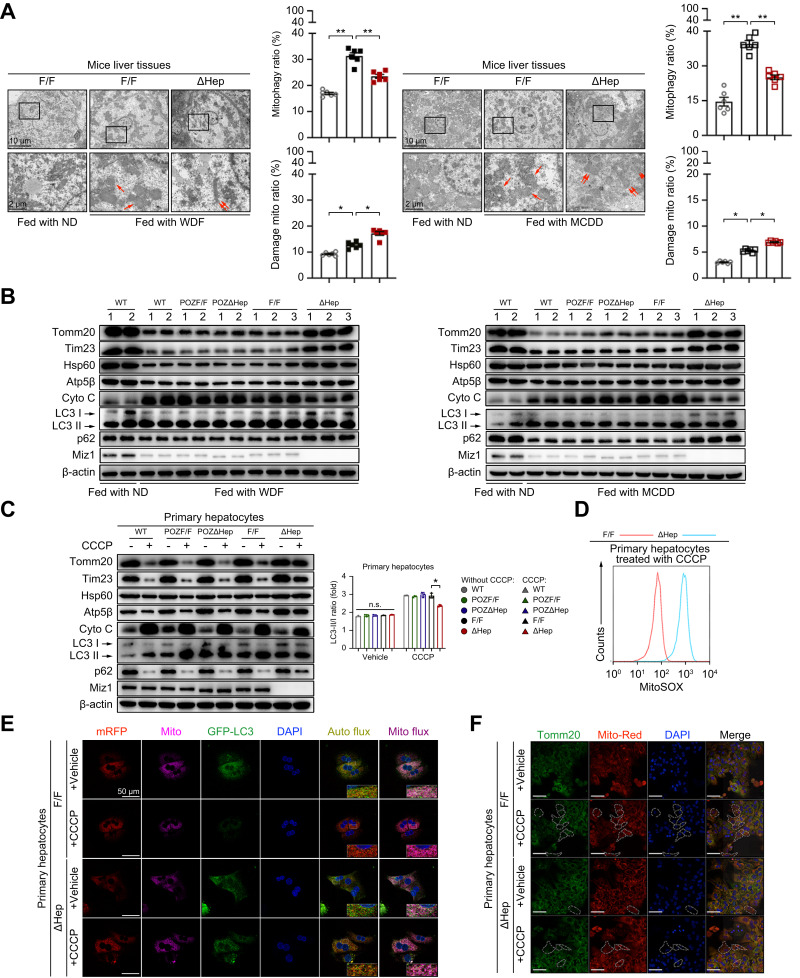
4. Miz1與PRDX6的(de)相互作用(yòng)阻斷了(le)PRDX6線粒體定位,增強了(le)線粒體泛素化(huà)
線粒體蛋白的(de)泛素化(huà)是其與自噬體膜結合的(de)關鍵步驟,免疫印迹顯示,Miz1缺失減少了(le)獨立于POZ結構域的(de)線粒體蛋白的(de)泛素化(huà)(圖4A, B)。爲了(le)證實Miz1如何調節線粒體泛素水(shuǐ)平,作者通(tōng)過免疫沉澱和(hé)質譜法穩定表達His-Miz1,确定了(le)HepG2細胞中可(kě)能與Miz1相互作用(yòng)的(de)蛋白(圖4C),并将PRDX6作爲一個(gè)有趣的(de)靶點(圖4D)。在F/F、ΔHep和(hé)POZΔHep原代肝細胞和(hé)過表達Miz1、Miz1(ΔPOZ)或PRDX6(以下(xià)簡稱HepG2-Miz1+、Miz1(ΔPOZ)+和(hé)PRDX6+)的(de)HepG2細胞中通(tōng)過共免疫沉澱證實了(le)這(zhè)種相互作用(yòng)(圖4E)。Miz1在肝細胞中獨立于POZ結構域與PRDX6相互作用(yòng)。
爲了(le)揭示Miz1、PRDX6與線粒體自噬之間的(de)關系,作者在過表達Miz1和(hé)/或PRDX6的(de)HepG2細胞以及感染慢(màn)病毒短發夾(sh)PRDX6的(de)F/F和(hé)ΔHep原代肝細胞中建立了(le)NASH模型。結果顯示,Miz1過表達導緻線粒體泛素增加,而PRDX6過表達在CCCP處理(lǐ)的(de)肝細胞中阻斷了(le)這(zhè)種作用(yòng)(圖4F)。免疫印迹顯示,CCCP處理(lǐ)後的(de)原代肝細胞和(hé)HepG2細胞中,PRDX6在MOM上的(de)定位程度比未治療時(shí)更大(dà)。此外,Miz1缺失導緻PRDX6的(de)線粒體定位增加,Miz1過表達導緻PRDX6在細胞質中積累,PRDX6在MOM上沒有定位,無論是否使用(yòng)CCCP(圖4G)。免疫熒光(guāng)也(yě)證實了(le)這(zhè)些事件(圖4H, I)。這(zhè)些結果表明(míng),Miz1作用(yòng)于PRDX6的(de)上遊,與PRDX6相互作用(yòng),阻斷PRDX6的(de)線粒體定位,從而增強線粒體泛素化(huà)和(hé)線粒體自噬。
此外,在MCDD誘導的(de)NASH模型中,AAV8-Miz1+、AAV8-PRDX6+和(hé)AAV8-雙過表達Miz1+和(hé)PRDX6+的(de)模型顯示,Miz1過表達減少而PRDX6過表達促進NASH進展。PRDX6過表達阻斷Miz1誘導的(de)NASH預防(圖4J)。TEM顯示Miz1過表達增強了(le)線粒體清除率,而PRDX6過表達可(kě)以阻斷這(zhè)種作用(yòng)(圖4J)。

5. Miz1降解導緻PRDX6解離并易位到Parkin Cys431,在NASH階段抑制Parkin自身泛素化(huà)和(hé)Parkin介導的(de)有絲分(fēn)裂,而在NAFLD階段則沒有。
WDF誘導NASH模型的(de)免疫沉澱顯示,Parkin在8周時(shí)與PRDX6相互作用(yòng),然後增加,而MG132阻斷了(le)這(zhè)種作用(yòng)(圖5A)。共免疫沉澱顯示Miz1、PRDX6和(hé)Parkin不能形成三蛋白複合物(wù)(圖5B)。接下(xià)來(lái),作者研究了(le)Parkin在PRDX6線粒體定位中的(de)作用(yòng)。免疫印迹顯示,Parkin缺失完全阻斷了(le)PRDX6的(de)線粒體定位(圖5C)。在線粒體自噬過程中,Parkin通(tōng)常是一種依賴于Cys431和(hé)Arg280的(de)泛素酶,共免疫沉澱顯示PRDX6在Cys431位點與Parkin相互作用(yòng),而在Arg280位點不相互作用(yòng)。
由于Parkin-Cys431在激活Parkin泛素酶活性中起作用(yòng),作者研究了(le)PRDX6與Parkin-Cys431相互作用(yòng)的(de)功能。Parkin Cys431突變抑制了(le)Miz1過表達誘導的(de)線粒體泛素化(huà)。Parkin Cys431突變和(hé)PRDX6過表達顯著抑制線粒體泛素化(huà)。同時(shí),PRDX6的(de)敲低恢複了(le)Miz1缺失導緻的(de)線粒體泛素減少,而Cys431的(de)突變阻斷了(le)PRDX6的(de)這(zhè)種功能(圖5D)。此外,AAV8-Miz1+/shPRDX6/Parkin+/Parkin-Cys431+被用(yòng)于MCDD誘導的(de)NASH模型。Miz1過表達和(hé)PRDX6敲低顯著抑制NASH進展,而Parkin-Cys431突變完全阻斷這(zhè)些作用(yòng)并增強NASH(圖5E)。此外,透射電鏡顯示,Miz1過表達和(hé)PRDX6敲低增強了(le)線粒體自噬,而Parkin-Cys431突變阻斷了(le)這(zhè)種作用(yòng)(圖5E)。這(zhè)些結果表明(míng),Miz1在NASH期間被泛素化(huà)降解。導緻PRDX6被募集到Parkin-Cys431,阻斷Parkin自泛素化(huà),從而抑制Parkin介導的(de)有絲分(fēn)裂。
最近的(de)一項研究表明(míng),PRDX6通(tōng)過抑制Notch對(duì)NAFLD具有保護作用(yòng),這(zhè)一結論與本文的(de)研究相反。因此,作者認爲PRDX6可(kě)能在NAFLD-NASH的(de)不同階段和(hé)不同刺激條件下(xià)具有雙重功能。首先,對(duì)還(hái)原性谷胱甘肽/氧化(huà)性谷胱甘肽比值的(de)分(fēn)析表明(míng),在飼喂WDF之前,PRDX6具有抗氧化(huà)作用(yòng)。此外,免疫印迹顯示,在飼喂WDF 8周後的(de)24小時(shí)FFA刺激中,PRDX6可(kě)以開始與Parkin相互作用(yòng)。同時(shí),PRDX6在該時(shí)間點之前動态升高(gāo),然後下(xià)降(圖5F-H)。這(zhè)些結果表明(míng),PRDX6在NAFLD階段具有有益的(de)抗氧化(huà)作用(yòng),但在NASH階段具有負性的(de)線粒體自噬抑制作用(yòng)。

6. 肝細胞中Miz1蛋白的(de)減少通(tōng)過PRDX6誘導的(de)Parkin介導的(de)線粒體自噬抑制,導緻NLRP3/IL-1β和(hé)NLRP3/TNFα軸的(de)激活增加
接下(xià)來(lái),作者在NASH小鼠模型中研究了(le)Miz1對(duì)炎症因子和(hé)炎症小體mRNA水(shuǐ)平的(de)影(yǐng)響。RT-qPCR結果顯示,Miz1缺失導緻炎性因子和(hé)炎性小體成分(fēn)mRNA表達增加。此外,在Miz1缺失組以及F/F和(hé)ΔHep原代肝細胞中,IL-1β、TNFα及其上遊炎性體NLRP3的(de)mRNA表達顯著升高(gāo)(圖6A)。然後,爲了(le)确認Miz1是否通(tōng)過增加線粒體自噬來(lái)調節NLRP3/IL-1β和(hé)NLRP3/TNF-α信号軸,作者用(yòng)抑制線粒體自噬的(de)藥物(wù)阻斷了(le)線粒體自噬。免疫印迹證實了(le)F/F和(hé)ΔHep原代肝細胞對(duì)線粒體自噬的(de)依賴抑制,并表明(míng)Miz1缺失導緻Caspase-1和(hé)NLRP3的(de)激活增加,而抑制線粒體自噬使這(zhè)些作用(yòng)惡化(huà)(圖6B)。然後,作者在體内用(yòng)liensinine23和(hé)Brefeldin A24阻斷線粒體自噬或用(yòng)MCC95025阻斷NLRP3的(de)激活(圖6C, D)。結果表明(míng),抑制線粒體自噬可(kě)阻斷Miz1過表達誘導的(de)NASH及IL-1β和(hé)TNFα分(fēn)泌抑制。此外,抑制NLRP3激活顯著阻斷Miz1缺陷誘導的(de)NASH加重和(hé)IL-1β和(hé)TNFα分(fēn)泌(圖6E, F)。ELISA在體外證實了(le)Miz1、線粒體自噬、NLRP3、IL-1β和(hé)TNFα分(fēn)泌之間的(de)關系(圖6G)。
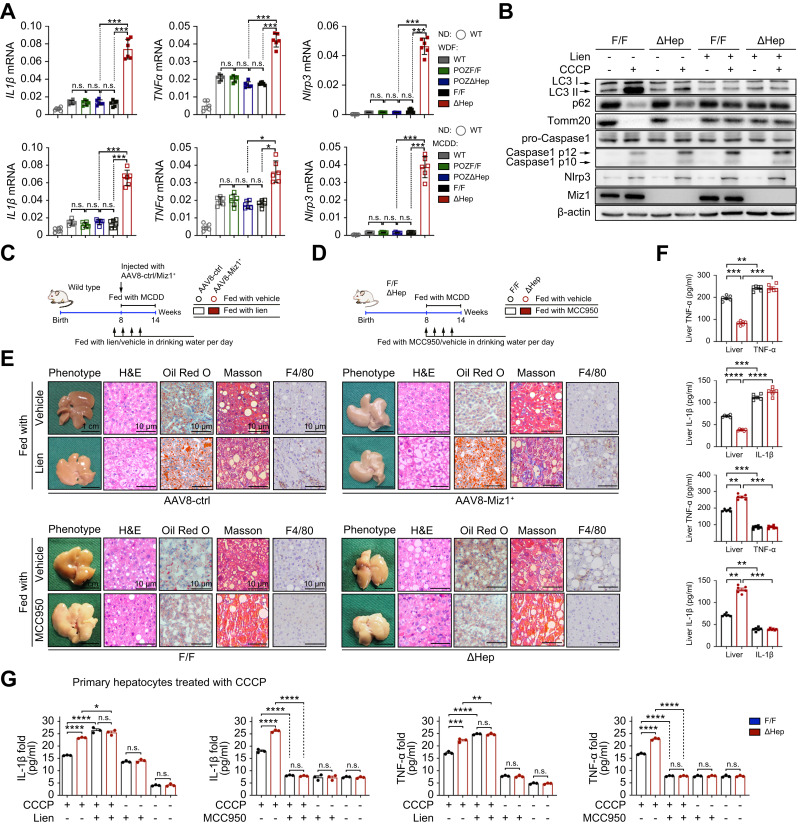
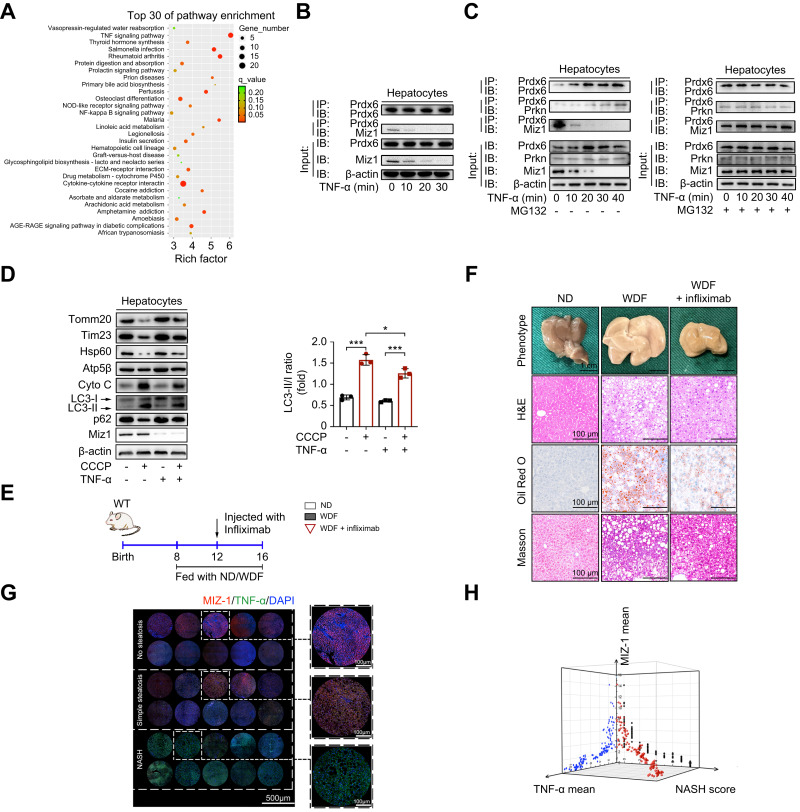
7. TNFα-誘導的(de)肝細胞Miz1蛋白減少可(kě)通(tōng)過PRDX6/Parkin軸抑制線粒體自噬
作者對(duì)正常和(hé)NASH組織的(de)RNA測序數據進行KEGG分(fēn)析顯示,NASH進展過程中TNF信号通(tōng)路高(gāo)度富集(圖7A),提示NASH肝髒中肝細胞Miz1蛋白的(de)減少可(kě)能是由肝髒炎症微環境自然引起的(de)。從WT小鼠中提取的(de)原代肝細胞分(fēn)别用(yòng)TNFα、IL-1β或IL-6處理(lǐ),免疫印迹顯示隻有TNFα誘導Miz1降解,而MG132阻斷Miz1降解。這(zhè)些結果表明(míng),Miz1可(kě)以通(tōng)過E3泛素化(huà)被TNFα特異性降解。
爲了(le)确定Miz1降解是否也(yě)會在NASH中引起類似的(de)分(fēn)子變化(huà),用(yòng)TNFα刺激WT原代肝細胞來(lái)模拟自然NASH條件下(xià)的(de)炎症環境。共免疫沉澱顯示,TNFα刺激減少了(le)與PRDX6相互作用(yòng)的(de)Miz1的(de)數量(圖7B)。當TNFα誘導Miz1降解時(shí),定位于MOM的(de)PRDX6數量增加,而用(yòng)MG-132抑制TNFα誘導的(de)Miz1降解可(kě)阻斷這(zhè)一作用(yòng)。此外,WT原代肝細胞的(de)共免疫沉澱顯示,随著(zhe)TNFα刺激,Parkin與PRDX6相互作用(yòng)的(de)數量增加,而MG132介導的(de)Miz1降解抑制阻止了(le)這(zhè)種作用(yòng)(圖7C)。
免疫印迹法顯示,TNFα抑制CCCP誘導的(de)WT原代肝細胞有絲分(fēn)裂(圖7D)。同時(shí),利用(yòng)可(kě)抑制TNFα功能的(de)英夫利昔單抗處理(lǐ)WDF誘導的(de)NASH。結果顯示,抑制TNFα可(kě)維持線粒體自噬,抑制炎症小體/炎症因子軸,從而限制NASH進展(圖7E, F)。共聚焦免疫熒光(guāng)檢測人(rén)類樣本中的(de)Miz1和(hé)TNFα,發現Miz1與人(rén)類樣本中NASH和(hé)TNFα水(shuǐ)平呈負相關(圖7G, H)。這(zhè)些結果表明(míng)脂肪肝中Miz1蛋白水(shuǐ)平與NAFLD的(de)嚴重程度和(hé)TNFα水(shuǐ)平呈負相關。
8.肝細胞中Miz1的(de)降低通(tōng)過類器官PRDX6/Parkin軸限制線粒體自噬,從而加重NASH進展
爲了(le)證實人(rén)類NASH的(de)這(zhè)些分(fēn)子機制,作者提取并構建了(le)肝類器官,并檢測了(le)這(zhè)些關鍵點。結果顯示,Miz1在NASH組中減少,導緻線粒體自噬和(hé)NLRP3減少,而過表達Miz1逆轉了(le)這(zhè)些變化(huà)(圖8A, B)。同時(shí),免疫印迹證實了(le)類器官中Miz1、線粒體自噬和(hé)NLRP3之間的(de)關系。共免疫沉澱揭示了(le)Miz1, PRDX6和(hé)Parkin在類器官中的(de)相關性(圖8C, D)。這(zhè)些結果表明(míng)這(zhè)些分(fēn)子機制存在于人(rén)肝髒組織中。

三、總結
總結一下(xià)這(zhè)篇文章(zhāng)的(de)主要發現:
1. NASH患者肝細胞Miz1水(shuǐ)平較低。
2. Miz1結合PRDX6并阻斷其抑制有絲分(fēn)裂的(de)能力。
3. 肝細胞Miz1的(de)減少降低了(le)線粒體自噬,增強了(le)肝細胞和(hé)巨噬細胞TNFα的(de)産生。
4. TNFα通(tōng)過E3泛素化(huà)進一步降低Miz1,導緻線粒體自噬進一步降低。
安徽省腫瘤防治所 安徽省腫瘤防治辦公室
Copyright © 2019 www.ahzlfzs.com,All rights reserved
版權所有 © 安徽腫瘤防治所 未經許可(kě) 嚴禁複制 備案号:
地址:合肥市蜀山區(qū)績溪路218号 電話(huà):0551-62922302